CHAPLE Syndrome (which stands for Complement Hyperactivation, Angiopathic Thrombosis, and Protein-Losing Enteropathy), also known as CD55 deficiency or DAF deficiency, is an exceptionally rare genetic disorder impacting the immune system. This article sheds light on the intricacies of CHAPLE Syndrome, highlighting its signs, genetic underpinnings, inheritance patterns, and potential treatments.
Understanding CHAPLE Syndrome
CHAPLE Syndrome, an acronym for “CD55 deficiency with hyper-activation of complement, angiopathic thrombosis, and severe protein-losing enteropathy (PLE),” is a genetic disorder that typically emerges during childhood. Initially described by Özen et al. in 2017, this condition poses significant health risks and complications.
Signs and Symptoms
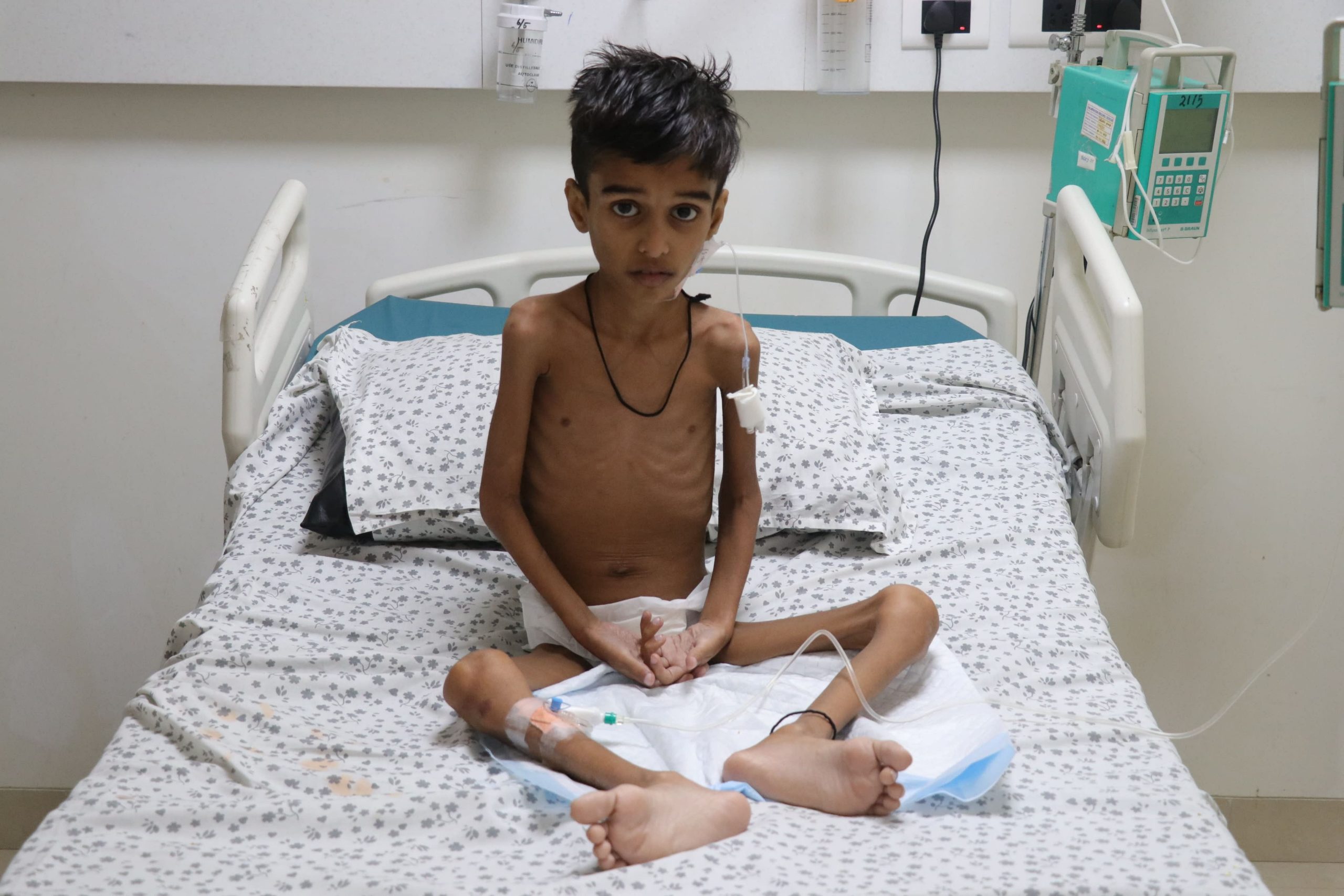
The hallmark of CHAPLE Syndrome is its severe protein-losing enteropathy, leading to hypoproteinemia. Common symptoms include abdominal discomfort, nausea, vomiting, diarrhea, loss of appetite, weight loss, and edema. Additionally, chronic malabsorption results in deficiencies of vital nutrients such as iron, ferritin, calcium, magnesium, folate, vitamin D, and vitamin B12. Some individuals with CHAPLE Syndrome experience recurrent respiratory infections due to hypogammaglobulinemia. Moreover, severe thrombotic vascular occlusions may also arise.
Genetic Basis
CHAPLE Syndrome stems from mutations in the CD55 gene, responsible for encoding the complement regulator protein. These mutations result in a loss of CD55 protein expression, triggering aberrant immune responses.
Inheritance Pattern
This syndrome is mainly inherited in an autosomal recessive manner, indicating that both parents contribute a mutated gene copy, leading to a homozygous defect in the child.
Pathophysiology
The central pathology of CHAPLE Syndrome revolves around complement-mediated autoimmune hemolysis and paroxysmal nocturnal hemoglobinuria. The CD55 protein, also known as the decay-accelerating factor, normally regulates the complement cascade’s amplification phase within the innate immune system. However, the absence of CD55 prompts the complement system to attack and destroy red blood cells, causing hemolysis.
Diagnosis and Treatment
CHAPLE Syndrome diagnosis typically combines clinical presentation, histological analysis, and genetic testing. While symptom presentation may vary, patients generally exhibit early-onset gastrointestinal symptoms, edema, malnutrition, hypoalbuminemia, and hypogammaglobulinemia. Histopathological examination of intestinal biopsy samples indicates primary intestinal lymphangiectasia. Large-vein thrombosis is also a potential concern. Treatment strategies are tailored to the individual’s condition. Promisingly, off-label use of eculizumab, an anti-C5 monoclonal antibody and complement inhibitor, has yielded positive outcomes in some patients over an 18-month period. Ongoing research efforts at institutions like Marmara University and the National Institute of Allergy and Infectious Diseases aim to further comprehend and address this complex disorder.
Conclusion
CHAPLE Syndrome, a rare genetic disorder with serious health implications, demands continued research to enhance diagnosis and treatment approaches. As medical science advances, a better understanding of CHAPLE Syndrome holds the promise of improving the lives of those affected by this intricate condition.